What the FPKM? A review of RNA-Seq expression units
This post covers the units used in RNA-Seq that are, unfortunately, often misused and misunderstood. I’ll try to clear up a bit of the confusion here.
The first thing one should remember is that without between sample normalization (a topic for a later post), NONE of these units are comparable across experiments. This is a result of RNA-Seq being a relative measurement, not an absolute one.
Preliminaries
Throughout this post “read” refers to both single-end or paired-end reads. The concept of counting is the same with either type of read, as each read represents a fragment that was sequenced.
When saying “feature”, I’m referring to an expression feature, by which I mean a genomic region containing a sequence that can normally appear in an RNA-Seq experiment (e.g. gene, isoform, exon).
Finally, I use the random variable 
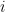
![\mathbb E[X_i]](https://s0.wp.com/latex.php?latex=%5Cmathbb+E%5BX_i%5D&bg=ffffff&fg=000000&s=0&c=20201002)
Counts
“Counts” usually refers to the number of reads that align to a particular feature. I’ll refer to counts by the random variable

where 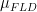
Since counts are NOT scaled by the length of the feature, all units in this category are not comparable within a sample without adjusting for the feature length. This means you can’t sum the counts over a set of features to get the expression of that set (e.g. you can’t sum isoform counts to get gene counts).
Counts are often used by differential expression methods since they are naturally represented by a counting model, such as a negative binomial (NB2).
Effective counts
When eXpress came out, they began reporting “effective counts.” This is basically the same thing as standard counts, with the difference being that they are adjusted for the amount of bias in the experiment. To compute effective counts:

The intuition here is that if the effective length is much shorter than the actual length, then in an experiment with no bias you would expect to see more counts. Thus, the effective counts are scaling the observed counts up.
Counts per million
Counts per million (CPM) mapped reads are counts scaled by the number of fragments you sequenced (


I’m not sure where this unit first appeared, but I’ve seen it used with edgeR and talked about briefly in the limma voom paper.
Within sample normalization
As noted in the counts section, the number of fragments you see from a feature depends on its length. Therefore, in order to compare features of different length you should normalize counts by the length of the feature. Doing so allows the summation of expression across features to get the expression of a group of features (think a set of transcripts which make up a gene).
Again, the methods in this section allow for comparison of features with different length WITHIN a sample but not BETWEEN samples.
TPM
Transcripts per million (TPM) is a measurement of the proportion of transcripts in your pool of RNA.
Since we are interested in taking the length into consideration, a natural measurement is the rate, counts per base (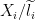

TPM has a very nice interpretation when you’re looking at transcript abundances. As the name suggests, the interpretation is that if you were to sequence one million full length transcripts, TPM is the number of transcripts you would have seen of type
I’m fairly certain TPM is attributed to Bo Li et. al. in the original RSEM paper.
RPKM/FPKM
Reads per kilobase of exon per million reads mapped (RPKM), or the more generic FPKM (substitute reads with fragments) are essentially the same thing. Contrary to some misconceptions, FPKM is not 2 * RPKM if you have paired-end reads. FPKM == RPKM if you have single-end reads, and saying RPKM when you have paired-end reads is just weird, so don’t do it :).
A few years ago when the Mortazavi et. al. paper came out and introduced RPKM, I remember many people referring to the method which they used to compute expression (termed the “rescue method”) as RPKM. This also happened with the Cufflinks method. People would say things like, “We used the RPKM method to compute expression” when they meant to say they used the rescue method or Cufflinks method. I’m happy to report that I haven’t heard this as much recently, but I still hear it every now and then. Therefore, let’s clear one thing up: FPKM is NOT a method, it is simply a unit of expression.
FPKM takes the same rate we discussed in the TPM section and instead of dividing it by the sum of rates, divides it by the total number of reads sequenced (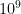
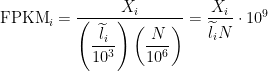
The interpretation of FPKM is as follows: if you were to sequence this pool of RNA again, you expect to see 

Relationship between TPM and FPKM
The relationship between TPM and FPKM is derived by Lior Pachter in a review of transcript quantification methods (
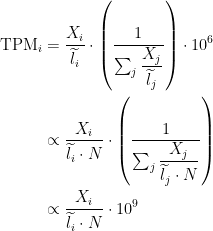
If you have FPKM, you can easily compute TPM:
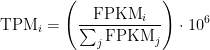
Wagner et. al. discuss some of the benefits of TPM over FPKM here and advocate the use of TPM.
I hope this clears up some confusion or helps you see the relationship between these units. In the near future I plan to write about how to use sequencing depth normalization with these different units so you can compare several samples to each other.
R code
I’ve included some R code below for computing effective counts, TPM, and FPKM. I’m sure a few of those logs aren’t necessary, but I don’t think they’ll hurt :).
countToTpm <- function(counts, effLen)
{
rate <- log(counts) - log(effLen)
denom <- log(sum(exp(rate)))
exp(rate - denom + log(1e6))
}
countToFpkm <- function(counts, effLen)
{
N <- sum(counts)
exp( log(counts) + log(1e9) - log(effLen) - log(N) )
}
fpkmToTpm <- function(fpkm)
{
exp(log(fpkm) - log(sum(fpkm)) + log(1e6))
}
countToEffCounts <- function(counts, len, effLen)
{
counts * (len / effLen)
}
################################################################################
# An example
################################################################################
cnts <- c(4250, 3300, 200, 1750, 50, 0)
lens <- c(900, 1020, 2000, 770, 3000, 1777)
countDf <- data.frame(count = cnts, length = lens)
# assume a mean(FLD) = 203.7
countDf$effLength <- countDf$length - 203.7 + 1
countDf$tpm <- with(countDf, countToTpm(count, effLength))
countDf$fpkm <- with(countDf, countToFpkm(count, effLength))
with(countDf, all.equal(tpm, fpkmToTpm(fpkm)))
countDf$effCounts <- with(countDf, countToEffCounts(count, length, effLength))

Very Cool!
Nice post!
What about the TMM? Is that also a within-sample normalization method?
Thanks for the comment. TMM is a between sample normalization, primarily used for comparing counts across numerous samples. I’ll talk about some of these methods in a post soon.
can you fix the link to the wagner paper? it’s down atm.
very helpful and the writing is crystal clear. thanks a bunch!
Thanks for the comment. The link seems to be working on my end. Here is the the PubMed:
http://www.ncbi.nlm.nih.gov/pubmed/22872506
Hi,
Great post and explanation. One thing that could be useful to clarify (at least for me):
TPM is normalized to the sum of the abundance of each transcripts, while FPKM is normalized to the number of reads sequenced.
However, as you know, not all sequenced reads map the genome, and not all mapped reads are assigned to a transcript.
I understand that the FPKM to TPM equation cancels out the normalization factor, but still, it’s never been clear for me if FPKM normalizes to “number of sequenced reads”, “number of mapped reads” or “number of assigned reads”. And from a practical stand point, I don’t see where Cufflinks gets the information on the number of sequenced reads if the input is a BAM file.
Thanks for the comment. Great question. I can’t speak for any of the engineers (Cufflinks, eXpress, etc.) as I’ve never worked on any of the abundance estimation software. I can say that intuitively I would normalize by the number of reads that are compatible with at least one transcript.
Although, at the end of the day if you’re comparing within a sample I don’t think it really matters, it will just change the scale of the number, but within a sample everything will be normalized by the same N. Between samples it is probably more important to just stay consistent (and make sure you adjust the N by one of many methods). Still, I don’t think it makes sense to use the absolute total as some of it could just be noise from the sample prep.
You are correct, though. I should definitely clarify. I guess it seems like everyone just says “the total number” when I think they mean “the total number of compatible reads.” I’ll ask around and update the post accordingly. The more I think about it though, I am more certain that N should be the total number of compatible reads.
If the input is a BAM file, you could in theory still store unmapped reads in the BAM file. (I think ‘samtools view -f 4 file.bam’ will give you the unmapped reads, assuming they are in the file). A lot of the mappers I’ve seen usually don’t report these, I imagine just to save disk space.
Great post! This is worthy of publication and indexing in Pubmed IMHO.
Extremely helpful. Thanks!
Thank you very much for the extremely useful information. One thing that puzzles me all the time. Say I’m doing an RNA-SEQ experiment where I’m comparing the effect of overexpression of a transcription factor to a control sample. Is differential expression the only way to determine the change in expression ? what I mean here is fold change and ratio. Normally people go for a 2 fold change cutoff to determine upregulation and downregulation (beside p-value and q-value). There are instances where you see FPKM changing from 0.1 to 1 which means in terms of differential expression that the upregulation is 10 folds. However, in other instances you see the FPKM value changing from 700 to 1000, in this case this gene that had 300 FPKM change is not considered upregulated because it did not pass the 2-fold change cutoff. Can we say in this case that this gene that jumped from 700 to 1000 is upregulated ? can we use the FPKM values to make plots and to show that a gene is affected by a treatment ? or the sole way to show upregulation is fold change ?
I would really love to know your opinion about that matter.
Thank you very much
Hi Waleed,
Thanks for your response and question.
I think the best “safe” way to detect whether a gene is upregulated is to perform a differential expression test with biological replicates. Such a test is typically formulated to check if the mean expression is different between two conditions (though there are other methods such as DESeq2 which looks at the fold change). In that case, if it comes back significant (with some caveats), I suppose I think it’s fair to say the gene is up/down regulated.
I’m generally not really a fan of specifying a particular cutoff as showing up/down regulation.
Thanks,
Harold
Thank you very much for the response Harold,
The reason why I asked that question is because I have been analyzing RNA-SEQ data for quite a while and I noticed that just using the FPKM ratio between test and control i.e. Log2(Test FPKM/control FPKM) can over/underestimate the significance of up/downregulation, exactly like the example I showed in the question. we normally do three replicates for each (Control and Test) and there is very little variation between replicates. I have tried both, the conventional Log2 ratio and tried as an example taking the difference between FPKM values (Average FPKM test – Average FPKM control). I found that the latter actually gives much better results than the conventional log2 (ratio). I’ve even test the output of the two methods with promoter motif analysis and gene ontology and the difference between FPKMs give much better results.
But that doesn’t mean that this difference between FPKMs is perfect. It has a problem of again deciding an appropriate cutoff for FPKM difference and that problem becomes very clear when looking at highly abundant transcripts. For example, if you set FPKM difference cutoff to ten then a gene that is expressed at 1000 FPKM and changes to 1010 would be marked as significantly upregulated though it is highly unlikely that a 10 FPKM increase/decrease would make a difference in the abundance of that transcript. I’m not an expert in anyway but I want to know if that way of looking at expression data is appropriate or if it is too good to be true.
One last request and sorry for asking too many question. Someone told me about a method to analyze RNA-SEQ data called Global Mean Normalization. have you head of it ? if so would you be willing to comment on that method
Thank you very much indeed Harold
Thank you for the post!
I am using GTEx RNA-seq data. How can I get the length of the genes in order to calculate TPM?
Sorry, My english is poor. I don’t understand effective length. May you give me example about it ? Thank you
he denominator is going to be different between experiments, and thus is also sample dependent which is why you cannot directly compare TPM between samples
thats simply wrong, ure using TPM BECAUSE you can compare it within samples and between experiments, the denominator is dependent on your annotation= #known transcripts, as long as u got the same Annotation among experiments it stays comparable. And even if u get different annotation the difference is still minor. Therefore see this paper. Please correct it in your blog-post! U can even see it int he Wagner paper
Hi Alex,
Thanks for your comment. Actually, the denominator is still dependent on the sample. Here is a simple counter example to your claim:
Now notice, that the total relative distribution has changed, but the only gene that likely changed it’s absolute expression is DEG. This is why you can’t compare TPMs directly.. TPM is a measurement of relative expression. If one gene changes relative expression, then all of the genes expressions change since they depend on the total expression rates.
I hope this clears things up.
Hi Harold,
DEG has 15 counts, so the total number of reads in experiment B is increased as well. It is not an appropriate comparison. You should assume that G1 has 1 read, G2 has 1 read, G3 has 1 read and DEG has 9 reads so that the total amount of reads remains the same. In this case, the dominator is the same if the genes are identical in length.
Woody
The counter example does not really apply to a more realistic situation, because in a real experiments we are typically working with thousands of genes, and the effect that you try to show with 4 genes is buffered. Let’s leave the example as above, except that you have 2100 genes instead of 4, a number that is still small but somewhat more to scale (E. coli has about 4000 genes; H. sapiens over 20000). Without the fancy formatting:
1- In experiment A the total number of counts is 3*2100 = 6300. Then, the TPM figure for each gene is 3/l divided by 6300/l, or 1/2100.
2- In experiment B the counts for all genes remain the same, except that DEG has 15 counts. Thus, the total number of counts is now 6312. Now, the TPM for all genes, except DEG, is 3/6312 or 1/2104, a change of less than 1% in expression (probably not significant). For DEG, however, we have 15/6312 or 5/2104, which is a nearly 5-fold increase, and likely significant.
The overall conclusion would be that the only gene that changed expression was DEG, by 5-fold, and expression of all other genes remained unchanged, the correct conclusion. In a real experiment, with genes of very different lengths and expression levels (and changes), the expectation is that the entire population, most of which we expect to show no change, has a buffering effect that protects against the case that you are trying to show.
In the experiment B, sequencing depth is increased twice. (12 reads -> 24 reads)
If gene G1, G2, and G3 still have 3 counts in the expr. B, it means those genes are less-expressed in the expr. B.
Very nice explanations here! But did I miss something or is there an error in the TPM equation?
According to the original RSEM paper ( bioinformatics.oxfordjournals.org/content/26/4/493.long ) the TPM equation does not start with
X_i / l_i but rather X_i / ( l_i * N ) where X_i = read counts, l_i = effective length and N = number of compatible reads in sample. That’s the same written by Lior Pachter in equation 10.
In the RSEM paper they state that t_i = v_i / l_i … where “we have that c_i/N approaches v_i” and v_i is the “fraction of nucleotides”.
Or did I misunderstand something here? (PS: Sorry for the ugly formula writing)
Ah ok, I think I figured it out: You have just canceled down the “N”.
Yup! That’s it.
I didn’t understand the effective length. It is hard to get the meaning of the sentence ‘Effective length refers to the number of possible start sites a feature could have generated a fragment of that particular length’. Could you explain the concept of the effective length more in detail?
This is the second time the question has been asked and not answered. Sorry, I can’t help you. I am also not sure what is being referred to as “effective length”.
Hello,
Thanks for this post. How do I calculate gene FPKM from its isoform FPKM values in eXpress? Can I just sum the isoform FPKM values to get gene FPKM?
Thanks,
Golsheed
Sorry for the late response. Yes, you can sum the FPKMs from isoforms to get gene FPKM.
Hi!
I am very new to R and RNAseq data, so I apologize if what I am asking is a little vague. I am working with TCGA Breast Cancer data and do not know how to check what kind of units my counts are in. How would I go about finding this information? I would like to use limma/edgeR as a statistical tool, but in reading the userguide I see that edgeR does not support FPKM/RPKM gene counts.
You’d have to check with the person who reported the expression table. If they are counts, then they are simply in counts of the number of times you saw a read from that feature. This is typically the space that most differential expression tools work in. They also often work in CPM.
Hi CW,
You may want to check out this table from the TCGA website: https://tcga-data.nci.nih.gov/tcga/tcgaDataType.jsp
It contains information about each of the data types deposited in their database. In particular, it says that the MAGE-TAB database associated with your dataset should contain information about how the expression values were calculated.
-Warren
I also remembered this other page on the TCGA wiki, which discusses the RNASeqV2 pipeline. There it reports that the values are RPKM: https://wiki.nci.nih.gov/display/TCGA/RNASeq+Version+2
(it links to the RNASeqV1 if that is how your data was generated)
Argh, I wish I could edit my comment. I misread the wiki. V1 uses RPKM, whereas V2 uses RSEM to quantify.
Hi, I’m just wondering if you ever wrote your post about “how to use sequencing depth normalization with these different units so you can compare several samples to each other”? This would be so useful. Thanks for a great explanation.
Hi Catherine,
The followup is here: https://haroldpimentel.wordpress.com/2014/12/08/in-rna-seq-2-2-between-sample-normalization/
Hope that helps,
Harold
Thanks Harold. This is a fantastic one-stop post on this topic.
I had a question for you. How does one account for RNASeq experiments that have a 3′ end bias? Specifically, in metrics that normalize the counts by the feature length, how does one handle 3′ bias. Do we just use the last exon length instead of the whole feature (gene/transcript)?
Thanks in advance for any help.
Hi,
I am also unclear on what effective length is. Can you please explain?
Also, if TPM cannot be used to compare data across experiments, is there any way to compare data from different RNA-Seq experiments?
Thank you!
Just for anyone else that is confused by this – here’s another good explanation of this:
https://groups.google.com/forum/#!topic/rsem-users/IaZmviqghJc
To make it easy, I’ll illustrate the concept of effective length with an example. Let’s say you are looking at a gene that is 100 nucleotides in length, and that each read is 30 exactly nucleotides long. Then, only reads starting at positions 1 through 71 in this gene can be found, because if you were to start sequencing at position 72 or further downstream then your read would be shorter than 30 (and you would not see it, if you follow the formula strictly). Thus, the effective length of this gene is 71 (because your target for sequencing is reduced to 71). Plugging the values in the formula, gene-length=100, mean-length-of-sequence-reads=30 (this is where it might become counterintuitive to some because there are surely sequence reads that are shorter, but this concept is statistic in nature, not strictly physical). Effective gene length is, then, 100-30+1 = 71.
I think I finally understood “effective length” thanks to your example 😉 Thanks for that!
Hi, is there any thing wrong with the statement that the FPKM_i is “basically just the rate of fragments per base multiplied by a big number (proportional to the number of fragments you sequenced) …”? From my understanding, here “big number” denotes the number of fragments “N”, right? If yes, it should be divided by it instead of “multiplied by” it.
Hi Harold, can I take TMM-normalized CPM values from edgeR, and simply divide by effective gene length to get TPM? Or is it more complicated?
Thanks!
Quick question: To go from FPKM to TPM, do you sum all the FPKMs of that gene’s transcripts within that sample or sum all the transcript’s FPKMs for all samples? If you had 3 transcripts and 2 samples. This is assuming one is working with between sample normalized FPKMs to start with.
if I want to calculate the TPM of a gene ,can I sum the TPM of the gene’s isoforms?
If I want to calculate a gene’s TPM ,can I sum the TPMs of all the gene’s isoforms?
Thanks for the post. Finally makes some sense and glad to have the math/references for once I’m actually calculating some of these things.
hi,
I would like ask the title of that Wagner et al paper at the end, beacuse I could not find where the link leads. Thanks!
Thank you! Now, I think i understood what FPKM really means.
Hi, I am starting to look at some RNA seq metadata and was struggling with the terminology and its interpretation and found your blog through some general search to look at terminology. I have to say that your post has been quite helpful, thank you.
Cheers, A
Hi Harold,
I have a question about the FPKM. I found out the FPKM range from 0 to 160000 in all the data I downloaded . Would you mind to explain what the values mean? For each dataset the gene expression value are majoriately between 0 and 1, but why there are some “outliers” here? Also considering about the up/down regulation, how can I know ?
Thanks,
Bill
Thanks for a good article.
I have a question in regarding to using one of several count metrics (RPKM, FPKM, TPM, etc) for doing ASE (allele specific expression). I think TPM might be a good choice since we are interested in calculating how many full length transcripts from each allele/haplotype align to a particular regions on the genome. But, I have not found any good article which discusses the statistics on doing counts analyses (allele A vs B) that align to the same gene. Plus, we are interested in doing ASE within each sample and next pool the counts from several samples to see how ASE changes in one sample vs. in a pool of samples.
Any suggestions?
Note the Wagner link mentioned above has moved, https://pdfs.semanticscholar.org/c98d/1c7ccb6b662c48403faedea967bd1d611d6c.pdf
Shouldn’t the sum of all TPM values of the same library equals one million?
Kallisto outputs a file named “abundance.tsv”, which contains tpm values for each transcript, but summing up these values won’t give one million. Why is that?
Many thanks!
Hey, nice article… is there any suggestion whereby you can perform BSN on FPKM values?
Hi! I have a quick question – when calculating the FPKM values should the total counts end up being less than 1 million? Thanks!
You can use ‘countToFPKM’ package.
This package provides an easy to use function to convert the read count matrix into FPKM matrix. It utilise the code in Trapnell, C. et al. (2010). The
fpkm()function requires three inputs to return FPKM as numeric matrix normalized by library size and feature length. This function takes read counts matrix of RNA-Seq data, feature lengths which can be retrieved using ‘biomaRt’ package, and the mean fragment lengths which can be calculated using the ‘CollectInsertSizeMetrics(Picard)’ tool. It then returns a matrix of FPKM normalised data by library size and feature effective length. It also provides the user with a reliable function to generate a FPKM heatmap plot of the highly variable features in RNA-Seq dataset.See https://github.com/AAlhendi1707/countToFPKM
Thank you a lot! When will your article about TMM normalization and co. appear?
Sorry, can anyone explain what does mean(FLD) mean? What is FLD?
As it reads above, “where \mu_{FLD} is the mean of the fragment length distribution which was learned from the aligned read.”